Publications & technical resources
Explore how DHO technology is facilitating scientific discovery

Thank you! Your submission has been received!
Oops! Something went wrong while submitting the form.
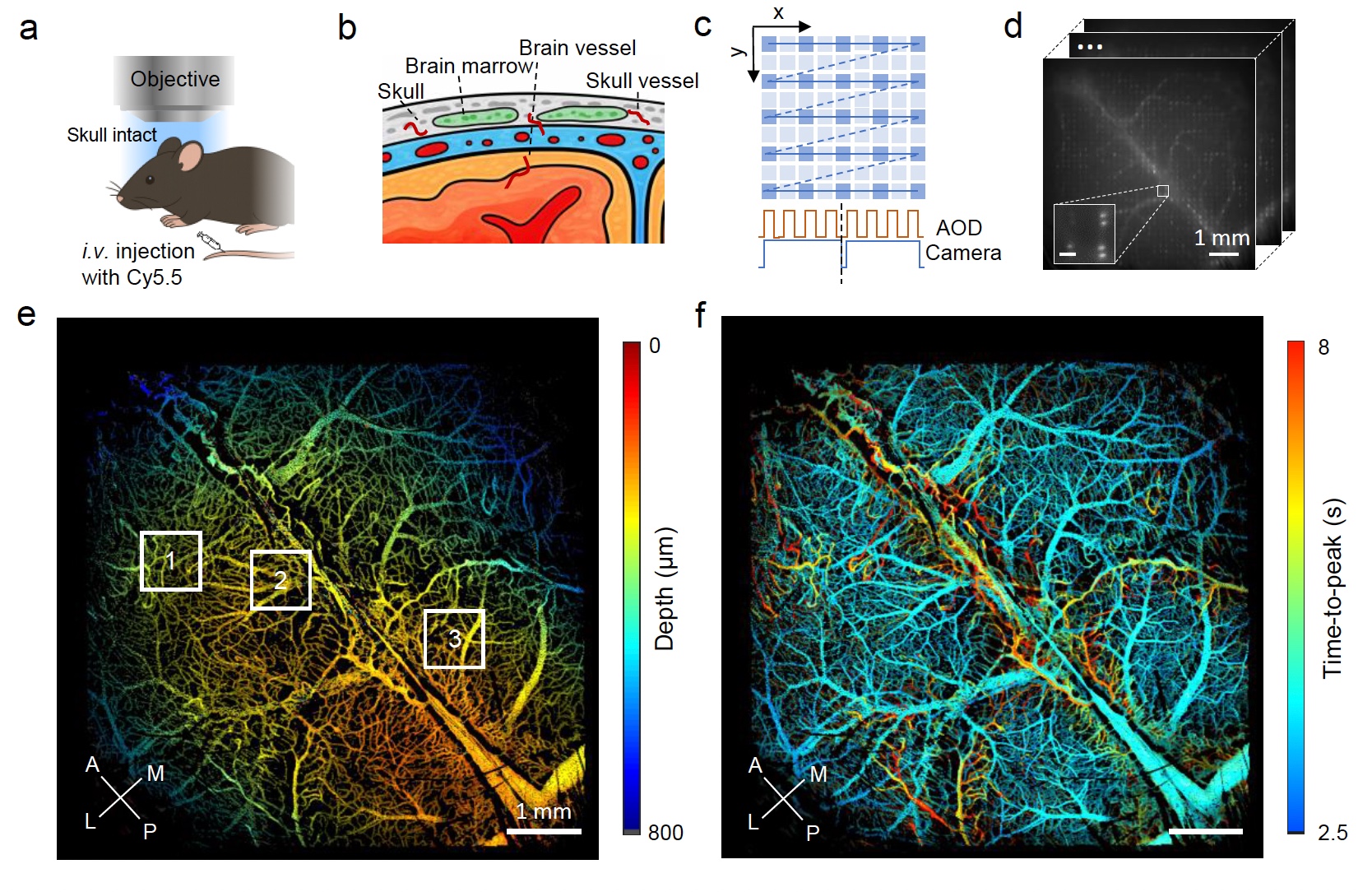
Double-helix optical point spread function enables real-time mesoscopic 3D functional microangiography in the living mouse brain and skull
Quantitative, volumetric imaging of cerebrovascular networks and microcirculation is essential for understanding brain function. However, rapid mesoscopic 3D imaging remains challenging because of fundamental trade-offs between spatiotemporal resolution, field of view, and sensitivity to functional parameters. Here we present a mesoscopic fluorescence imaging platform featuring a double-helix phase mask for real-time, depth-resolved measurements through the intact mouse skull. The compact phase-mask design is compatible with both laser-scanning and widefield microscopy. Using multifocal laser scanning, we demonstrate real-time volumetric in vivo imaging while discriminating calvarial from cerebral vasculature across 6.6×6.6×0.8 mm3 volume. Beyond high-resolution structural imaging, perfusion time-to-peak values are extracted from the laser-scanning configuration while accurate flow velocity/direction information is provided via widefield tracking of fluorescently labeled cells. We demonstrate the platform’s capabilities by analyzing brain-layer-specific perfusion dynamics and vascular topology in glioma-bearing mouse brains, offering unprecedented views for probing cerebrovascular alterations in both physiological and pathological contexts.
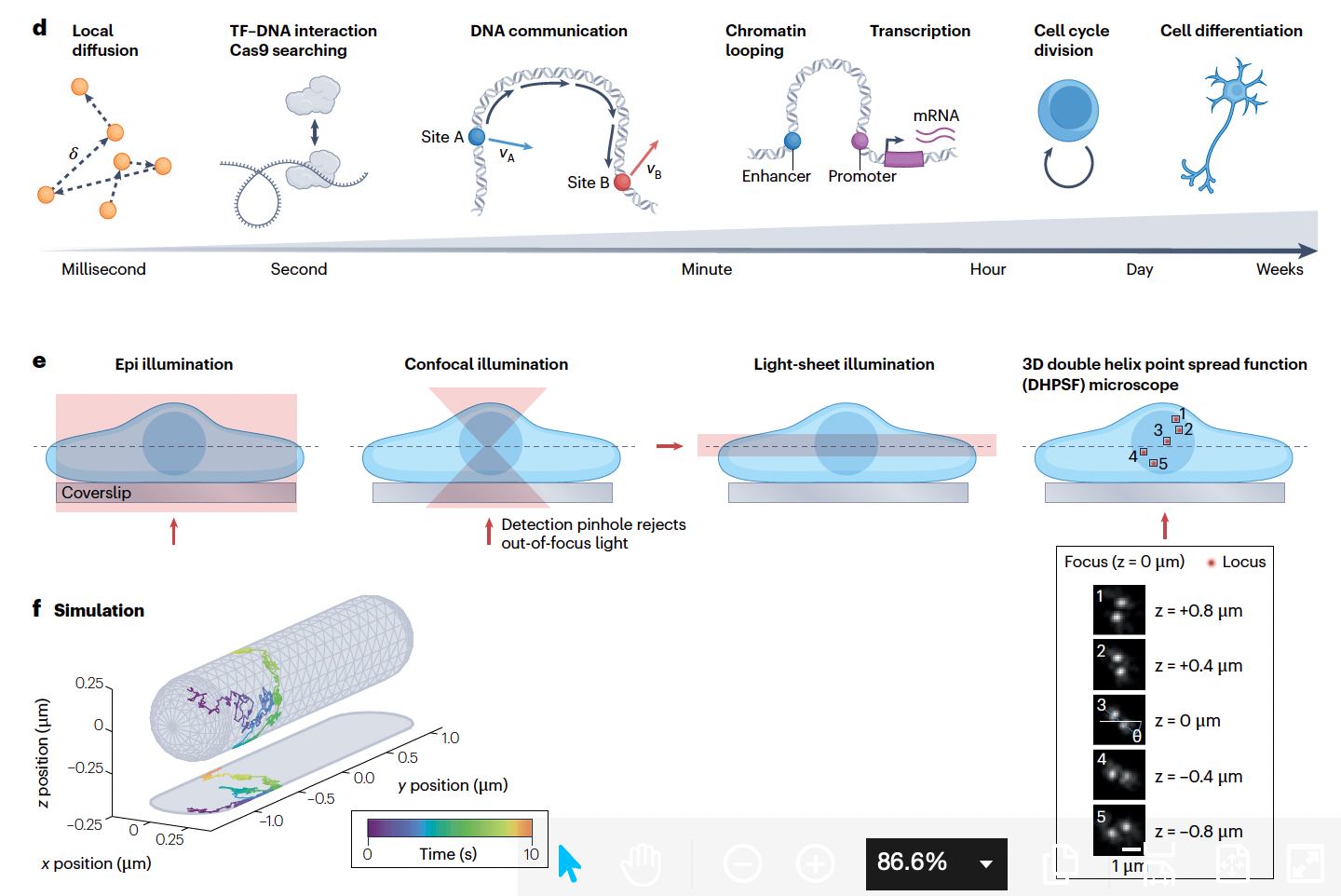
CRISPR–Cas-based live cell imaging of genome dynamics
The 3D architecture and dynamics of the genome are crucial for regulation of genome stability, transcription and cellular function. CRISPR-based live imaging technologies have enabled real-time visualization of specific genomic loci and transcripts in living cells. These tools harness customized guide RNAs and nuclease-deactivated Cas effectors to achieve precise genomic targeting, and recent methodological advances provide the 3D spatiotemporal resolution required to decipher real-time chromatin communication. These methods are elucidating the biophysical properties of chromatin, linking dynamic enhancer–promoter interactions directly to transcription, and revealing the role of 3D genome dynamics in basic cellular processes and disease. Here, we summarize the development of CRISPR-based live-cell imaging techniques, highlight the complementary 3D microscopy and analysis methods compatible with these methods, and offer perspectives on their applications to uncover fundamental principles that govern genome dynamics and function.

High-resolution dynamic imaging of chromatin DNA communication using Oligo-LiveFISH
Three-dimensional (3D) genome dynamics are crucial for cellular functions and disease. However, real-time, live-cell DNA visualization remains challenging, as existing methods are often confined to repetitive regions, suffer from low resolution, or require complex genome engineering. Here, we present Oligo-LiveFISH, a high-resolution, reagent-based platform for dynamically tracking non-repetitive genomic loci in diverse cell types, including primary cells. Oligo-LiveFISH utilizes fluorescent guide RNA (gRNA) oligo pools generated by computational design, in vitro transcription, and chemical labeling, delivered as ribonucleoproteins. Utilizing machine learning, we characterized the impact of gRNA design and chromatin features on imaging efficiency. Multi-color Oligo-LiveFISH achieved 20-nm spatial resolution and 50-ms temporal resolution in 3D, capturing real-time enhancer and promoter dynamics. Our measurements and dynamic modeling revealed two distinct modes of chromatin communication, and active transcription slows enhancer–promoter dynamics at endogenous genes like FOS. Oligo-LiveFISH offers a versatile platform for studying 3D genome dynamics and their links to cellular processes and disease.

A hierarchical pathway for assembly of the distal appendages that organize primary cilia
Distal appendages are nine-fold symmetric blade-like structures attached to the distal end of the mother centriole. These structures are critical for formation of the primary cilium, by regulating at least four critical steps: ciliary vesicle recruitment, recruitment and initiation of intraflagellar transport (IFT), and removal of CP110. While specific proteins that localize to the distal appendages have been identified, how exactly each protein functions to achieve the multiple roles of the distal appendages is poorly understood. Here we comprehensively analyze known and newly discovered distal appendage proteins (CEP83, SCLT1, CEP164, TTBK2, FBF1, CEP89, KIZ, ANKRD26, PIDD1, LRRC45, NCS1, C3ORF14) for their precise localization, order of recruitment, and their roles in each step of cilia formation. Using CRISPR-Cas9 knockouts, we show that the order of the recruitment of the distal appendage proteins is highly interconnected and a more complex hierarchy. Our analysis highlights two protein modules, CEP83-SCLT1 and CEP164-TTBK2, as critical for structural assembly of distal appendages. Functional assay revealed that CEP89 selectively functions in RAB34+ ciliary vesicle recruitment, while deletion of the integral components, CEP83-SCLT1-CEP164-TTBK2, severely compromised all four steps of cilium formation. Collectively, our analyses provide a more comprehensive view of the organization and the function of the distal appendage, paving the way for molecular understanding of ciliary assembly.

Whole-cell multi-target single-molecule super-resolution imaging in 3D with microfluidics and a single-objective tilted light sheet
Multi-target single-molecule super-resolution fluorescence microscopy offers a powerful means of understanding the distributions and interplay between multiple subcellular structures at the nanoscale. However, single-molecule super-resolution imaging of whole mammalian cells is often hampered by high fluorescence background and slow acquisition speeds, especially when imaging multiple targets in 3D. In this work, we have mitigated these issues by developing a steerable, dithered, single-objective tilted light sheet for optical sectioning to reduce fluorescence background and a pipeline for 3D nanoprinting microfluidic systems for reflection of the light sheet into the sample and for efficient and automated solution exchange. By combining these innovations with PSF engineering for nanoscale localization of individual molecules in 3D, deep learning for analysis of overlapping emitters, active 3D stabilization for drift correction and long-term imaging, and Exchange-PAINT for sequential multi-target imaging without chromatic offsets, we demonstrate whole-cell multi-target 3D single-molecule super-resolution imaging with improved precision and imaging speed.

Long-axial-range double-helix point spread functions for 3D volumetric super-resolution imaging
Single-molecule localization microscopy (SMLM) is a powerful tool for observing structures beyond the diffraction limit of light. Combining SMLM with engineered point spread functions (PSFs) enables 3D imaging over an extended axial range, as has been demonstrated for super-resolution imaging of various cellular structures. However, super-resolving structures in 3D in thick samples, such as whole mammalian cells, remains challenging as it typically requires acquisition and post processing stitching of multiple slices to cover the entire sample volume or more complex analysis of the data. Here, we demonstrate how the imaging and analysis workflows can be simplified by 3D single-molecule super-resolution imaging with long-axial-range double-helix (DH)-PSFs. First, we experimentally benchmark the localization precisions of short- and long-axial-range DH-PSFs at different signal-to-background ratios by imaging fluorescent beads. The performance of the DH-PSFs in terms of achievable resolution and imaging speed was then quantified for 3D single-molecule super-resolution imaging of mammalian cells by DNA-PAINT imaging of nuclear lamina protein lamin B1 in U-2 OS cells. Furthermore, we demonstrate how the use of a deep-learning-based algorithm allows the localization of dense emitters, drastically improving the achievable imaging speed and resolution. Our data demonstrate that using long-axial-range DH-PSFs offers stitching-free, 3D super-resolution imaging of whole mammalian cells, simplifying the experimental and analysis procedures for obtaining volumetric nanoscale structural information.
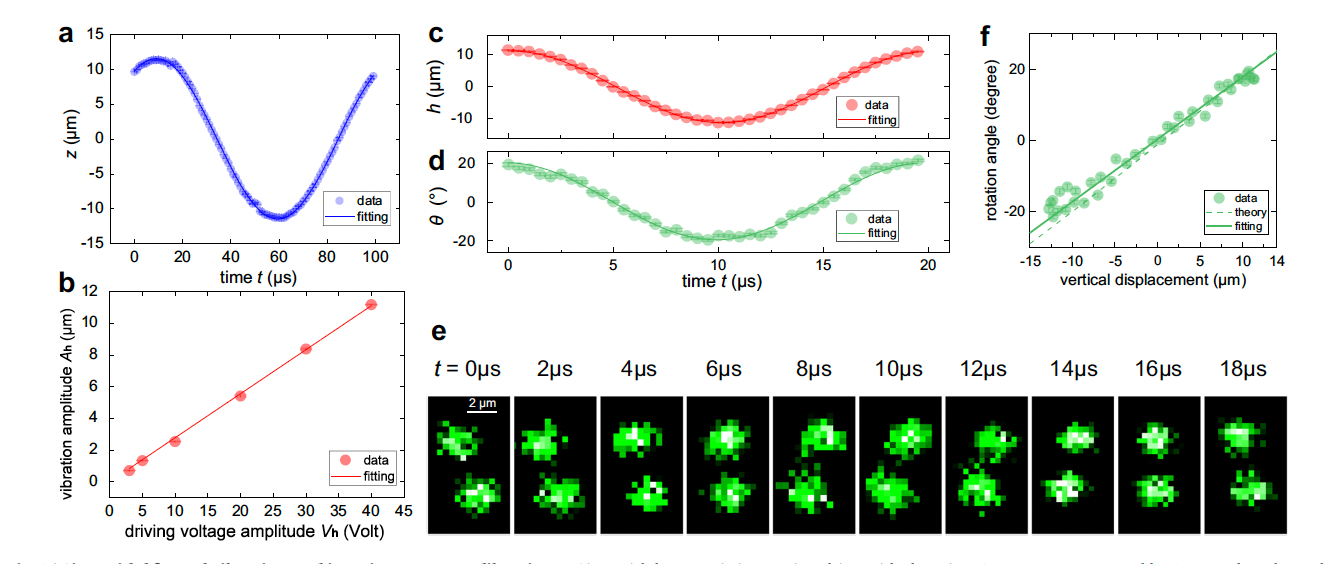
Tracking the extensive three-dimensional motion of single ions by an engineered point-spread function
Three-dimensional (3D) imaging of individual atoms is a critical tool for discovering new physical phenomena and developing new technologies in microscopic systems. However, the current single-atom-resolved 3D imaging methods are limited to static circumstances or a shallow detection range. Here, we demonstrate a generic dynamic 3D imaging method to track the extensive motion of single ions by exploiting the engineered point-spread function (PSF). We show that the image of a single ion can be engineered into a helical PSF, thus enabling single-snapshot acquisition of the position information of the ion in the trap. A preliminary application of this technique is demonstrated by recording the 3D motion trajectory of a single trapped ion and reconstructing the 3D dynamical configuration transition between the zig and zag structures of a 5-ion crystal. This work opens the path for studies on single-atom-resolved dynamics in both trapped-ion and neutral-atom systems.

Multimodal illumination platform for 3D single-molecule super-resolution imaging throughout mammalian cells
Single-molecule super-resolution imaging is instrumental in investigating cellular architecture and organization at the nanoscale. Achieving precise 3D nanometric localization when imaging structures throughout mammalian cells, which can be multiple microns thick, requires careful selection of the illumination scheme in order to optimize the fluorescence signal to background ratio (SBR). Thus, an optical platform that combines different wide-field illumination schemes for target-specific SBR optimization would facilitate more precise 3D nanoscale studies of a wide range of cellular structures. Here, we demonstrate a versatile multimodal illumination platform that integrates the sectioning and background reduction capabilities of light sheet illumination with homogeneous, flat-field epi- and TIRF illumination. Using primarily commercially available parts, we combine the fast and convenient switching between illumination modalities with point spread function engineering to enable 3D single-molecule super-resolution imaging throughout mammalian cells. For targets directly at the coverslip, the homogenous intensity profile and excellent sectioning of our flat-field TIRF illumination scheme improves single-molecule data quality by providing low fluorescence background and uniform fluorophore blinking kinetics, fluorescence signal, and localization precision across the entire field of view. The increased contrast achieved with LS illumination, when compared with epi-illumination, makes this illumination modality an excellent alternative when imaging targets that extend throughout the cell. We validate our microscopy platform for improved 3D super-resolution imaging by two-color imaging of paxillin – a protein located in the focal adhesion complex – and actin in human osteosarcoma cells.
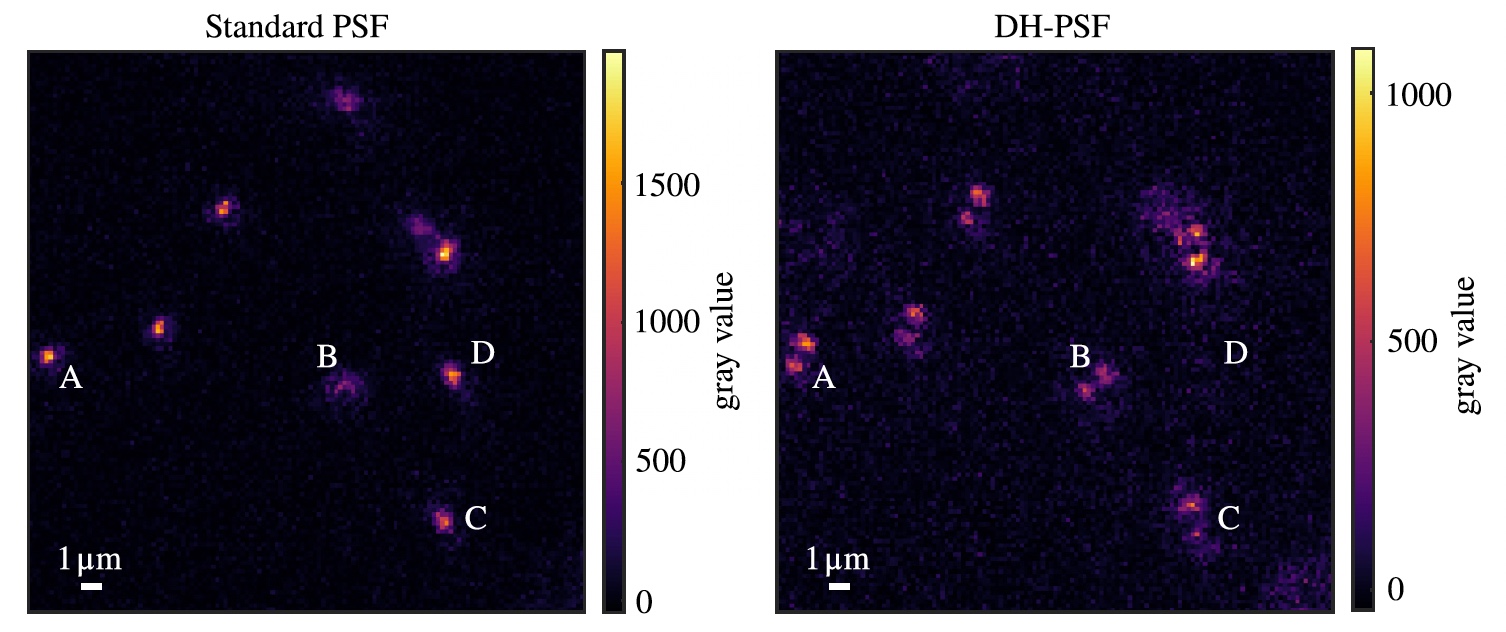
Three-dimensional imaging of single atoms in an optical lattice via helical point-spread-function engineering
We demonstrate a method for determining the three-dimensional location of single atoms in a quantum gas microscopy system using a phase-only spatial light modulator to modify the point-spread function of the high-resolution imaging system. Here, the typical diffracted spot generated by a single atom as a point source is modified to a double spot that rotates as a function of the atom’s distance from the focal plane of the imaging system. We present and numerically validate a simple model linking the rotation angle of the point-spread function with the distance to the focal plane. We show that, when aberrations in the system are carefully calibrated and compensated for, this method can be used to determine an atom’s position to within a single lattice site in a single experimental image, extending quantum simulation with microscopy systems further into the regime of three dimensions.
No results found
Please try different keywords
Thank you! Your submission has been received!
Oops! Something went wrong while submitting the form.









No results found
Please try different keywords
Thank you! Your submission has been received!
Oops! Something went wrong while submitting the form.
No results found
Please try different keywords